Cardiac amyloidosis in the 21st century

Heart failure
Cardiac amyloidosis is a relatively rare condition that has several different causes. Management involves pharmaceutical treatment of heart failure, which is fundamentally related to diastolic dysfunction, and treatment of the underlying disease process. Evolving treatments that target the source of amyloid proteins are dramatically improving the outlook for patients. Early diagnosis is vital for the best results to be achieved.
- The systemic amyloidoses are a group of disorders caused by deposition of misfolded insoluble proteins (amyloid) in different tissues and organs.
- Cardiac amyloidosis does not develop in all patients with systemic amyloidosis.
- Patients with cardiac amyloidosis present with heart failure (with preserved ejection fraction), fatigue and arrhythmia.
- Findings on echocardiography or cardiac MRI are distinctive and may direct further investigation.
- Technetium pyrophosphate bone scanning may help in diagnostic differentiation of the subtype of amyloidosis.
- Infiltration of amyloid protein in the interstitial spaces of the myocardium causes predominantly diastolic dysfunction. Diuretics are the mainstays of treatment, and modern heart failure medications may be harmful in this setting.
- Prognosis depends on ability to treat the underlying disease process.
Amyloid is formed when naturally occurring (soluble) ‘starch-like’ proteins that normally fold into specific molecular shapes instead misfold into pleated sheets that aggregate into insoluble fibrils.1-3 Amyloidosis is a systemic pathology, in which these insoluble fibrillar amyloid proteins are deposited in tissues and organs. The systemic amyloidoses share a common pathophysiological mechanism where these protein aggregates have toxic effects on native cells and disturb the interstitial structure of involved organs.
Many different amyloid proteins are recognised as causing cardiac infiltration but in practical terms they can be divided into two groups:
- proteins produced by the marrow
- proteins produced by the liver.
The marrow disorders are clonal plasma cell dyscrasias, in which a rogue population of a single plasma cell line produces immunoglobulin light chain proteins. In some cases, these monoclonally produced proteins will fold into the amyloid pattern. If the clonal plasma cells occupy more than 10% of the marrow then the condition qualifies as multiple myeloma. Smaller clonal percentages in the marrow represent the lower end of this spectrum and are referred to as light chain (AL) amyloidosis (previously known as primary amyloidosis).
The liver conditions associated with amyloidosis produce the protein transthyretin (TTR, previously known as prealbumin), which folds to become TTR amyloid (ATTR). There are two subsets of ATTR amyloid conditions. Some families have a gene mutation that creates a defective TTR molecule. Most cases of ATTR amyloidosis, however, are sporadic, with normal (‘wild-type’) transthyretin produced in excess. This condition is typically seen in older men (previously referred to as senile cardiac amyloidosis).
Very rarely, inflammatory processes (such as osteomyelitis or rheumatoid arthritis) may produce serum amyloid A (AA) and subsequent AA amyloidosis (e.g. autoimmune disease, chronic infection).3
Clinical manifestations
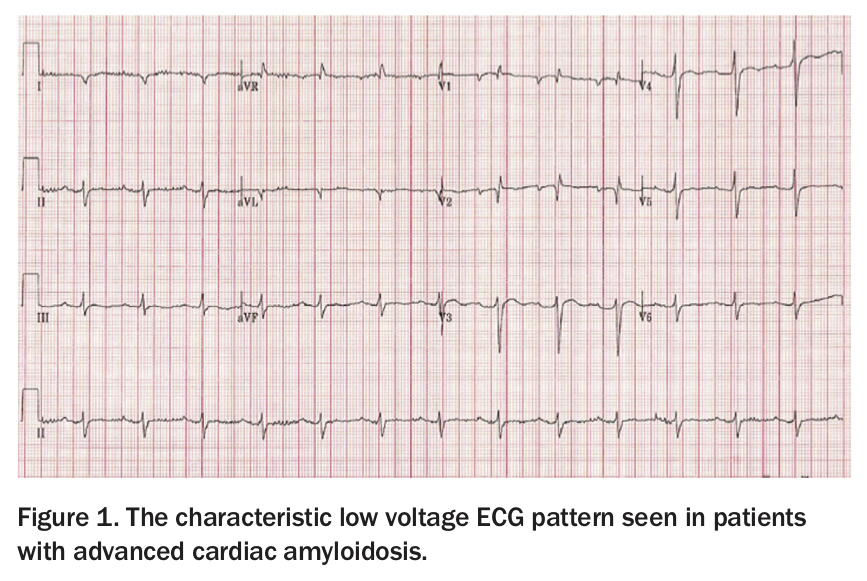
Presentations of systemic amyloidosis include dyspnoea (restrictive cardiomyopathy), renal involvement (up to a nephrotic picture), peripheral neuropathy, liver involvement, macroglossia, purpura and ecchymoses, and spontaneous bleeding.3 Cardiac infiltration (which is not present in all cases) presents with heart failure (with preserved ejection fraction), fatigue and arrhythmia. Despite the fact that the ventricular walls are thickened (often massively), the ECG classically shows low voltages (Figure 1) and/or atrial fibrillation.3 This is because the chamber walls contain the amyloid protein in the interstitial spaces of the myocardium, and the myocyte mass is actually decreased. The findings on echocardiography or cardiac MRI are distinctive, and these are frequently the initial tests that lead to further investigations and diagnosis.3
{kind=link}
Investigations
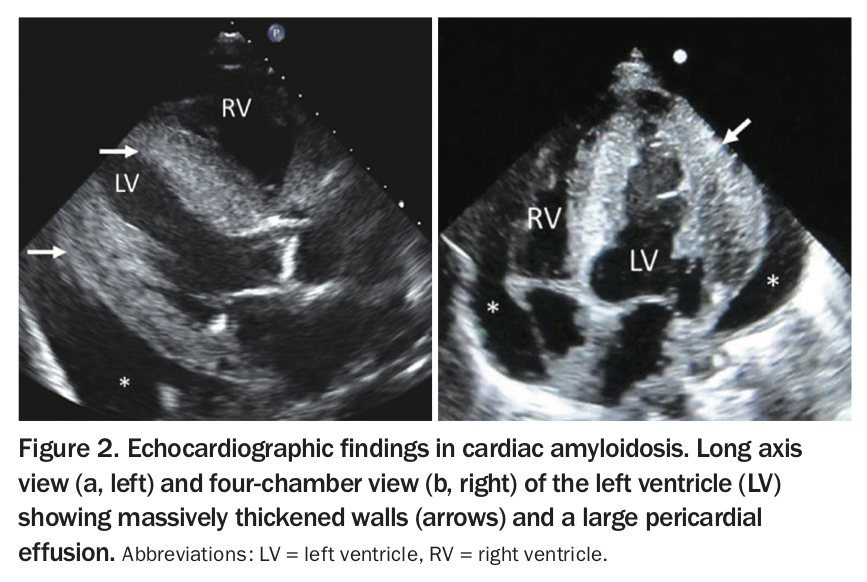
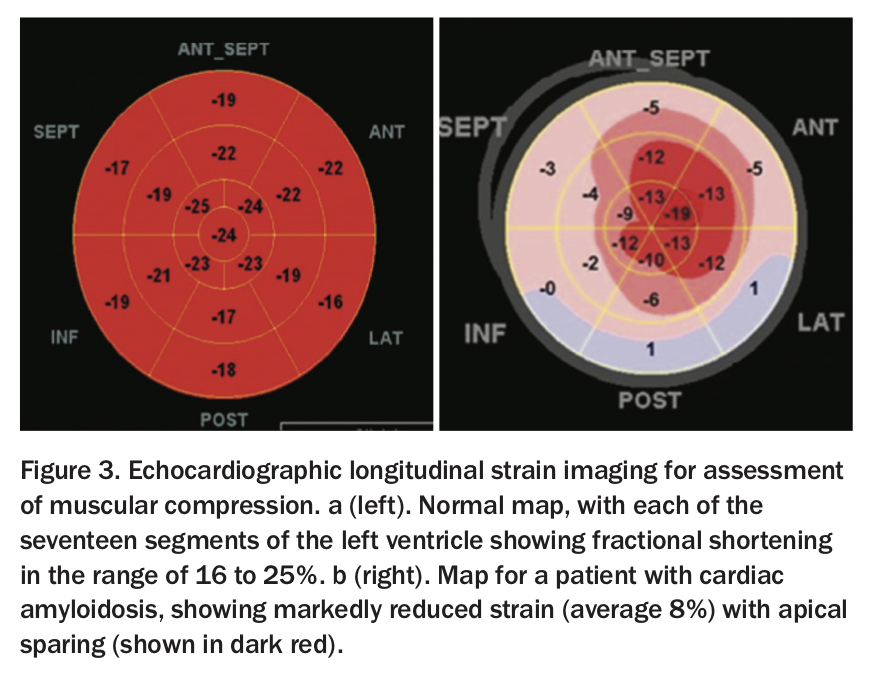
All types of cardiac amyloidosis have similar appearance on echocardiography. It is visually characteristic: thickened, speckled left and right ventricular walls, small ventricular cavities and large atria, with or without small pericardial effusions and advanced diastolic dysfunction (Figure 2).4,5 Global longitudinal strain imaging, which uses echocardiographic pixel tracking to assess muscular compression in the left ventricle, has revolutionised this diagnosis (Figure 3a). In patients with amyloid infiltration, this measure of cardiac efficiency is dramatically reduced in the base and mid-ventricle, with the classically described ‘apical-sparing’ pattern (Figure 3b).6,7
{kind=link}
{kind=link}
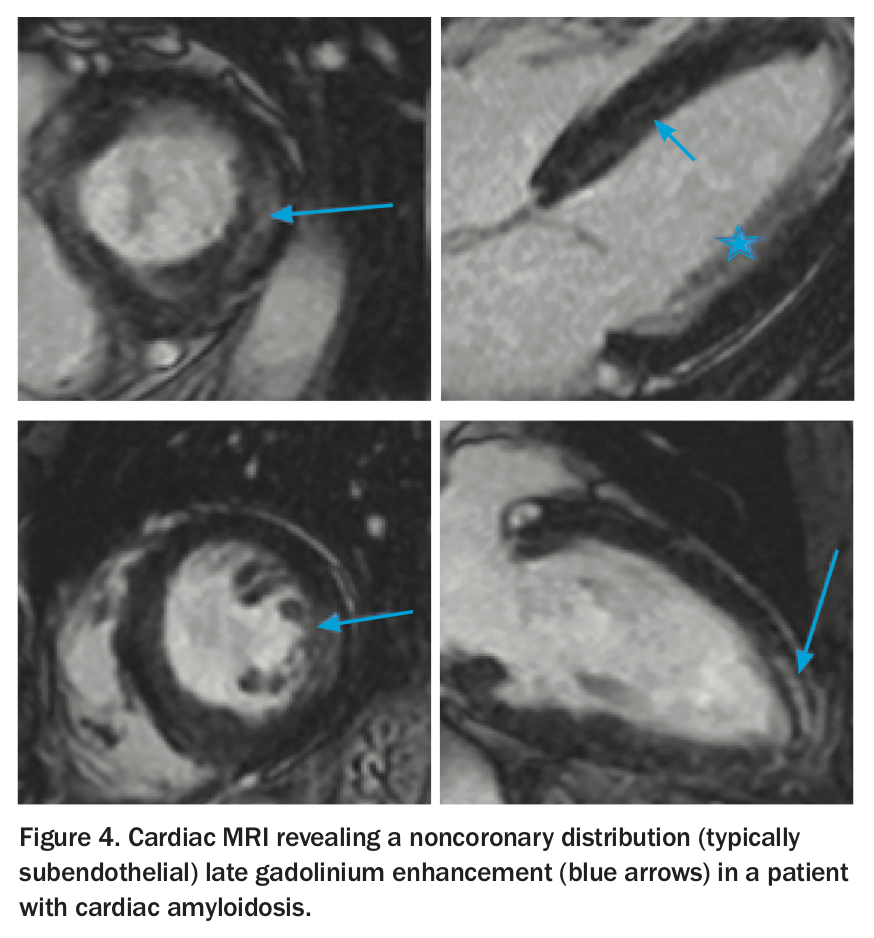
Cardiac MRI reveals a noncoronary distribution (typically subendothelial) late gadolinium enhancement, with difficulty nulling the myocardium (Figure 4).3,8 It is useful in further confirming the diagnosis.
{kind=link}
Laboratory tests that may be used include measuring brain natriuretic peptide and cardiac troponin levels (evidence of cardiac involvement). The marrow conditions (AL amyloidosis and multiple myeloma) are investigated with serum and urine electrophoresis, looking for monoclonal bands, and bone marrow biopsy.3 Tissue biopsy is generally recommended to confirm the amyloid proteins histologically which may be found in the marrow or peripheral organs such as the gut, kidney or skin or the fat of the abdominal wall.3
Recent studies have shown the unexpected utility of technetium pyrophosphate bone scans in ATTR cardiac amyloidosis. By amazing coincidence, there is marked uptake of this tracer in the heart in patients with ATTR amyloidosis, but not in those with AL amyloidosis or myeloma conditions (Figures 5a and b).3,9 Many patients can avoid cardiac biopsy if their imaging is consistent with the clinical picture and a diagnosis can be made after biopsy from another organ.10
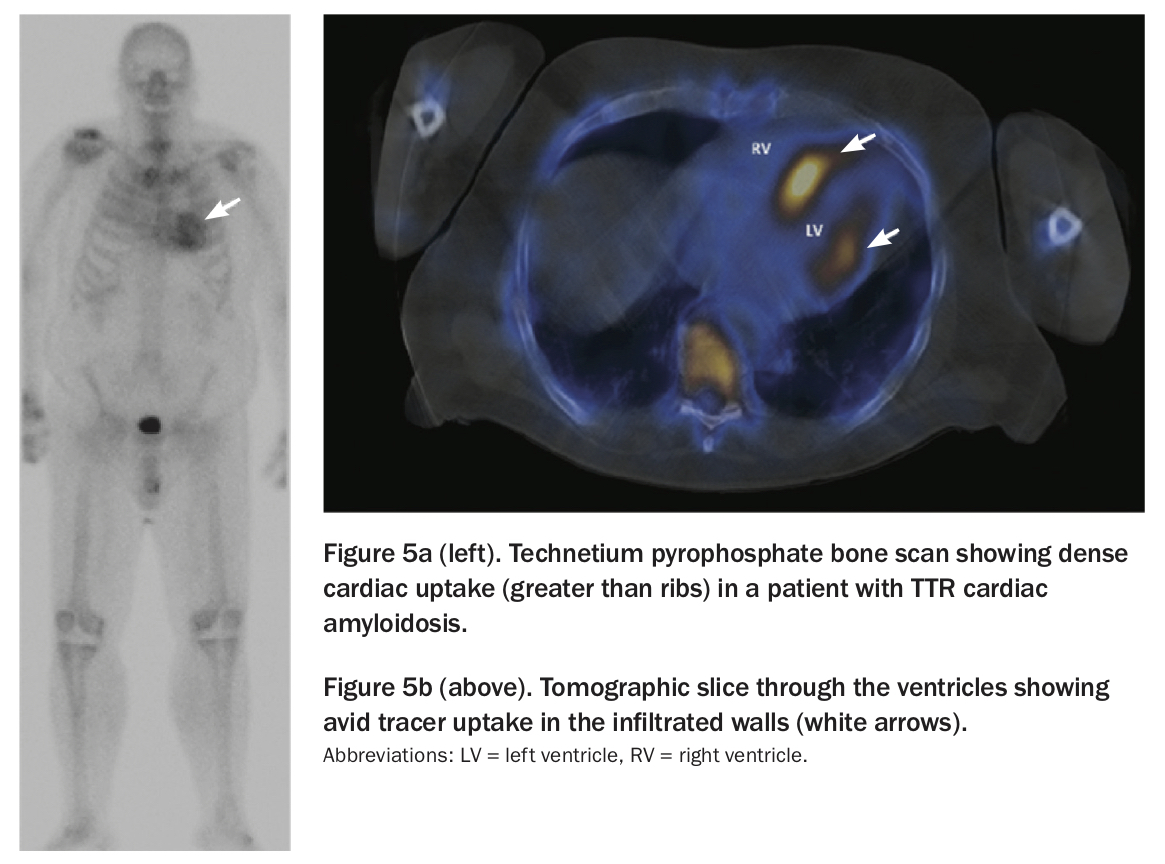{kind=link}
Management
The management of patients with cardiac amyloidosis involves treatment of heart failure and of the underlying disease. Diuretics are the mainstays of this treatment. Paradoxically, modern heart failure medications are often harmful in this setting. Due to the amyloid infiltration causing predominantly diastolic dysfunction, blood pressure is usually low and patients rarely tolerate renin-angiotensin-aldosterone system inhibitors. Beta blockers inhibit heart rate and prevent compensatory mechanisms in patients with amyloidosis, resulting in worsening heart failure. Digitalis tends to accumulate in amyloid tissue and increases risk of toxicity.3 Combinations of loop diuretics, thiazides and aldosterone antagonists often produce the best results.
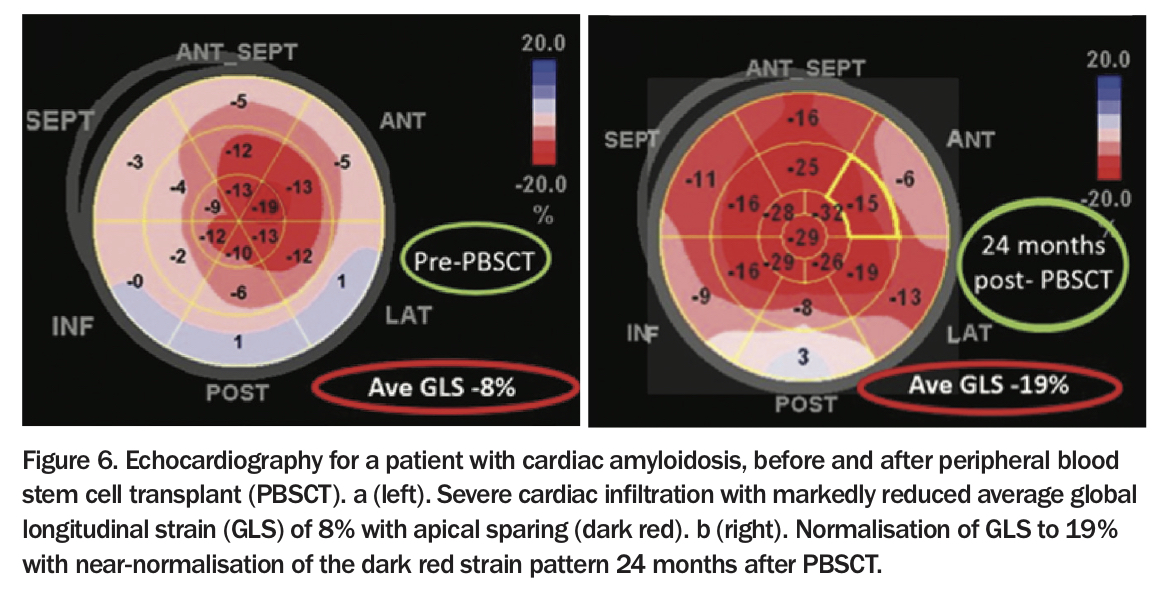
Historically, patients with untreated advanced AL amyloidosis or multiple myeloma had a very poor prognosis – median survival of 12 months, reduced to five months in the presence of cardiac involvement.1,2 In the mid-1990s, high dose chemotherapy with melphalan and peripheral blood stem cell transplantation showed that haematological remission could be achieved in selected patients, with median survival extending to over four years.11,12 When light chain production ceases, cardiac amyloid regression can be achieved, with further increases in life expectancy seen (Figure 6).10,13 Subsequently, a variety of antiplasma cell therapies, reflecting the introduction of immunomodulatory agents (e.g. thalidomide and lenalidomide), and protease inhibitors (e.g. bortezomib), have been used in the induction phase. Newer agents such as monoclonal antibodies and antiserum amyloid P antibodies are being investigated.
{kind=link}
For elderly patients with wild-type (non-familial) ATTR conditions, novel agents such as diflunisal (an NSAID) and monoclonal antibodies are in trials. Tafamidis has been shown to reduce mortality and hospitalisation and to improve functional capacity and quality of life in patients with ATTR amyloidosis.14 The prognosis of ATTR amyloidosis is better than for AL amyloidosis. Patients with wild-type ATTR amyloidosis can survive over six years from diagnosis.15 Survival for familial ATTR amyloidosis has been documented between two and 20 years (10-year survival rates between 30 and 80%).16 Liver transplantation may be offered for patients with some familial forms of ATTR amyloidosis.
Conclusion
Cardiac amyloidosis is a relatively rare condition that has several different causes. Its presentation can be variable. Imaging is characteristic, but the condition needs to be considered for the diagnosis to be made. Potential treatments are available for each of the different types, and early recognition is vital for improved survival. There is hope for patients with these potentially deadly diseases. CT
COMPETING INTERESTS: None.
References
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.