Pulmonary hypertension. What you need to know

Pulmonary hypertension
Pulmonary hypertension comprises a diverse group of conditions that lead to increased right ventricular afterload. Management and prognosis vary according to the cause. Patients at high risk (e.g. those with systemic sclerosis) or with unexplained progressive dyspnoea should be screened with echocardiography. Early referral to a specialist centre is key to timely investigation and management.
- The prognosis and management of pulmonary hypertension vary, depending on the underlying cause.
- Diagnosis relies on systematic evaluation of patients with dyspnoea and screening of those at high risk (e.g. patients with systemic sclerosis, congenital heart disease or previous pulmonary embolism).
- Specialist referral centres provide access to individualised work-up and management for patients; early referral is key to optimising outcome.
- Novel drug therapies and pulmonary vascular interventions are available for specific subgroups of pulmonary hypertension, such as pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension; however, for most patients, treatment targets the underlying condition.
Pulmonary hypertension (PH) refers to an aetiologically diverse group of conditions that result in abnormally elevated pressures in the pulmonary arterial bed, with consequent increased right ventricular afterload. The clinical manifestations vary widely, ranging from asymptomatic and well compensated in the early stages to progressive right heart failure and ultimately death.
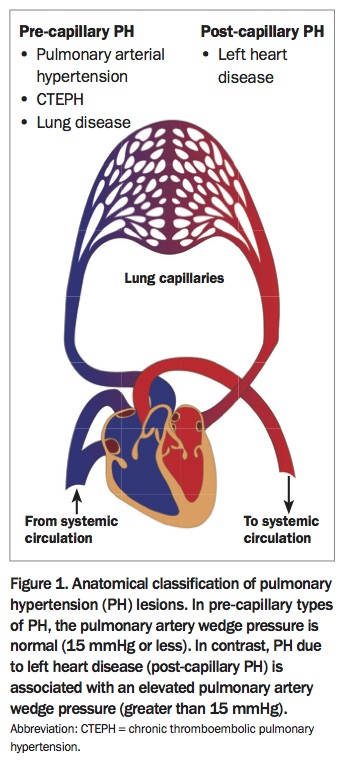
PH is defined by an elevation of the mean pulmonary artery pressure of 25mmHg or more at rest, measured on right-heart catheterisation. It is important to appreciate that PH is simply a description of the haemodynamic state of the pulmonary circulation, and many diseases and mechanisms can lead to elevated pulmonary artery pressure. From a haemodynamic perspective, PH is classified as:
- pre-capillary PH, when the site of disease is in the pulmonary arteries
- post-capillary PH, when the disease is due to an elevation in pulmonary venous pressure (Figure 1).
{kind=link}
Clinical classification of pulmonary hypertension
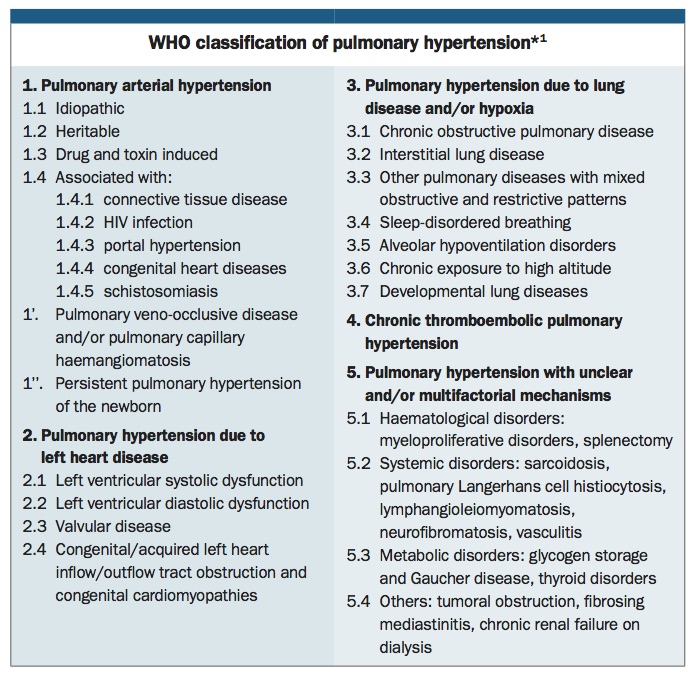
PH has been classified by the WHO into five groups based on similar disease pathogenesis, clinical presentation and therapeutic approach (Box).1 Identifying the underlying aetiology of PH can be a complex process. However, it is crucially important as both management options and overall prognosis vary substantially depending on the aetiology.
{kind=link}
Group 1. Pulmonary arterial hypertension
The pathobiology of pulmonary arterial hypertension (PAH) is complex, but the key feature is endothelial dysfunction leading to an imbalance of vasoactive mediators resulting in excessive vasoconstriction. Furthermore, the distal pulmonary arteries exhibit proliferative vascular remodelling, inflammation and thrombosis, causing irreversible narrowing.2 This results in increased resistance of the pulmonary arteries, with the finding of pre-capillary PH (i.e. on the arterial side of the pulmonary capillary bed) on right-heart catheterisation. Aetiologically, PAH can be:
- idiopathic
- associated with a connective tissue disease (most commonly systemic sclerosis)
- associated with congenital heart disease with a left-to-right shunt (such as atrial and ventricular septal defects)
- secondary to a drug or toxin (e.g. anorexigens, methamphetamine and dasatinib)
- secondary to a germline mutation.
Group 2. Pulmonary hypertension due to left-sided heart disease
Left heart disease is the most common cause of PH. In this group, PH arises through passive transmission of elevated left atrial pressure to the pulmonary artery. It is also known as post-capillary PH or pulmonary venous hypertension (i.e. on the venous side of the pulmonary capillary bed). Diseases that elevate left atrial pressure include mitral and aortic valve disease and conditions that result in stiffening of the left ventricle (diastolic dysfunction), such as hypertension, aortic stenosis, hypertrophic cardiomyopathy, ischaemic heart disease and infiltrative conditions such as amyloid cardiomyopathy. Pulmonary venous hypertension can lead to secondary remodelling of the pulmonary arteries, an initially reactive process that may become irreversible over time.
Group 3. Pulmonary hypertension due to lung disease or chronic hypoxia
Chronic obstructive pulmonary disease (COPD), interstitial lung diseases, obstructive sleep apnoea, obesity hypoventilation syndrome and other related conditions may lead to PH via several mechanisms. These include destruction of the functional alveolar capillary bed, chronic hypoxic vasoconstriction, inflammation and the mechanical stress of hyperinflated lungs. Similarly, some individuals exposed to chronically low partial pressures of oxygen at altitude will develop pulmonary vascular remodelling and PH.
Group 4. Chronic thromboembolic pulmonary hypertension
In chronic thromboembolic pulmonary hypertension (CTEPH), elevated pulmonary pressure is predominantly secondary to nonresolving pulmonary thromboembolism that occludes the pulmonary arteries and undergoes organisation and fibrosis. This results in the formation of thrombotic intra-arterial ‘webs’ and irreversible remodelling of the pulmonary arteries.
Group 5. Pulmonary hypertension with multifactorial or unclear mechanisms
In this group, the pathobiology of PH may be multifactorial or unclear.
Epidemiology
The exact prevalence of PH in the general community is unclear as population-based studies are scarce. A Western Australian study of patients referred for outpatient echocardiography found a prevalence of PH of about 9%, with women affected almost twice as commonly as men.3 PH secondary to left heart disease is the most common group. Up to two-thirds of patients with systolic and diastolic left ventricular dysfunction and symptomatic aortic stenosis have secondary PH on transthoracic echocardiography (TTE), as well as almost all patients with severe symptomatic mitral stenosis.4
The prevalence of PH in patients with lung disease such as COPD or interstitial lung disease varies depending on the severity of the underlying lung disease. In patients being assessed for lung transplantation for severe respiratory disease, the prevalence of PH has been reported as about 25%.5 However, PH in patients with severe lung disease is usually only of mild to moderate severity. Nevertheless, a small proportion (about 5 to 10%) may develop severe PH.
Following acute pulmonary embolism, 0.5 to 4% of patients may develop CTEPH, with major risk factors for PH including large, recurrent or unprovoked emboli.6 However, there is no evidence that thrombolytic therapy used to treat acute large pulmonary embolism prevents the future development of CTEPH.7
PAH by contrast is a rare but often severe disease, with a reported prevalence of 15 to 50 cases per million.8 Prevalence is high in certain patient groups, occurring in:
- up to 10 to 15% of patients with systemic sclerosis (also known as scleroderma), irrespective of subtype
- up to 10% of patients with congenital heart disease
- 2 to 6% of patients with portal hypertension
- 0.5% of patients with HIV infection.
International guidelines recommend annual screening for PH with echocardiography in patients with systemic sclerosis.4
The prevalence of PH is rising in developed countries, because of an increasing prevalence of underlying conditions such as obstructive sleep apnoea, COPD and diastolic heart disease, as well as increased awareness and diagnosis.9
Clinical features
The clinical hallmark of PH is a history of progressive exertional dyspnoea. This is often disproportionate to the impairments in gas exchange and respiratory mechanics. As the disease progresses, worsening pulmonary perfusion results in less effective gas exchange and low cardiac output due to right ventricular failure, with consequent hypoxaemia and exercise limitation.
Rising pulmonary pressures increase right ventricular afterload, leading to elevated right heart pressures and the clinical features of right heart failure. These include peripheral oedema, ascites, hepatic congestion and renal impairment due to renal venous congestion. In advanced disease, patients may experience arrhythmias and syncope.
When PH is suspected based on clinical features or is detected on TTE, the clinical features of possible underlying conditions should be sought. These include symptoms and signs of connective tissue diseases (e.g. Raynaud’s phenomenon, skin thickening, inflammatory joint and muscle diseases, gut dysmotility), pulmonary disease, obstructive sleep apnoea and left heart disease.
Investigations
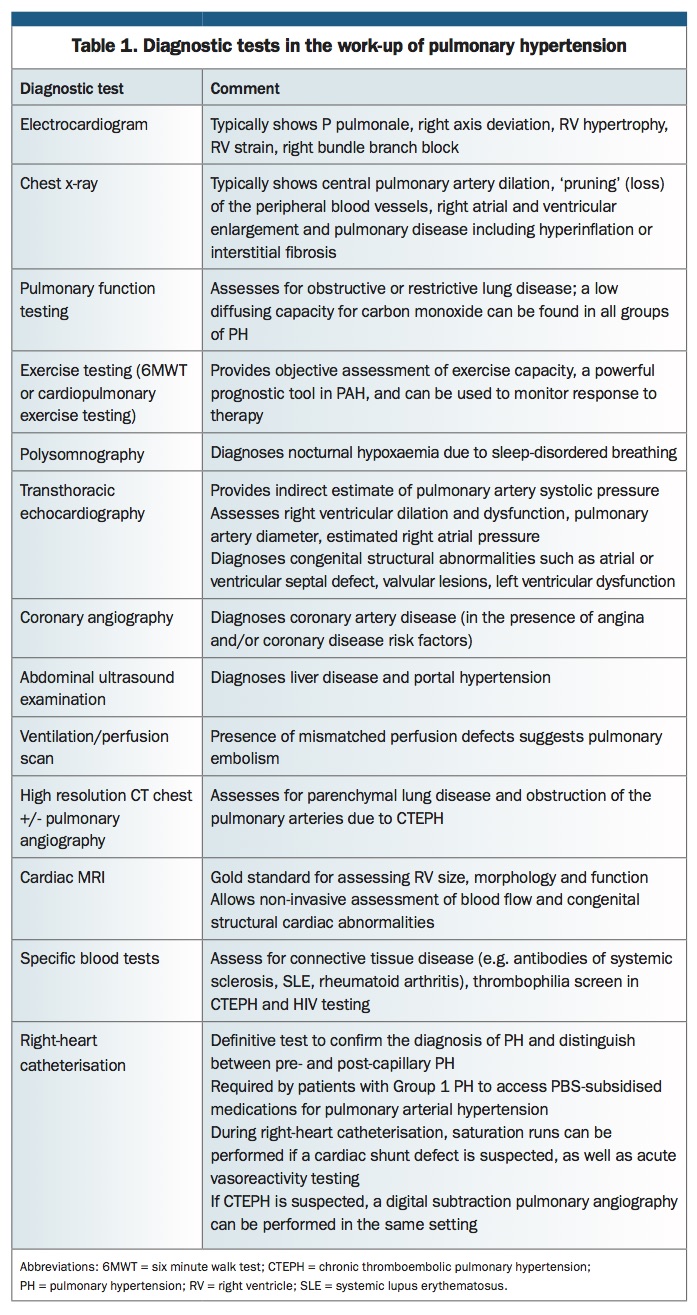
An overview of diagnostic tests that may be relevant in patients with PH is shown in Table 1.
{kind=link}
Transthoracic echocardiography
TTE is the most useful screening test when pulmonary hypertension is suspected. Findings on TTE in patients with PH include:
- elevated pulmonary artery pressure estimated from the tricuspid regurgitation jet velocity
- right ventricular dilation and impairment
- inferior vena cava dilation caused by elevated right atrial pressure.
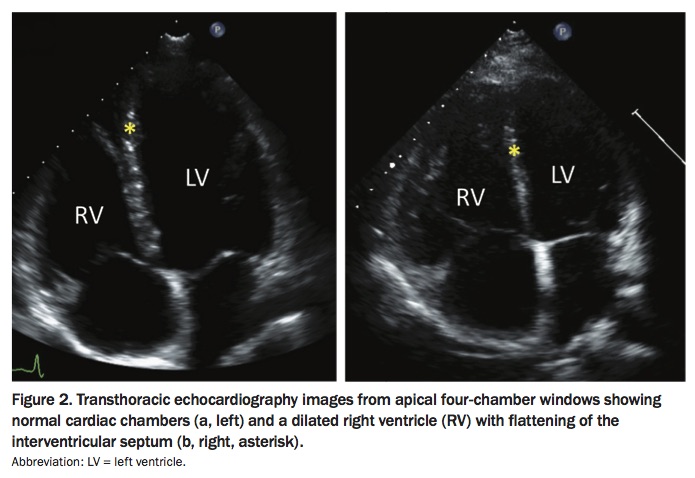
TTE can also provide important information on whether PH is due to left heart disease, for example by showing the presence of left heart valvular disease or left ventricular dysfunction (Figure 2). Right ventricular function needs to be carefully assessed on TTE, as right heart function is the main determinant of prognosis in PH.
{kind=link}
Diagnostic work-up
After identification of PH, the diagnostic work-up is directed at confirming the diagnosis and determining the cause (Flowchart). In some cases, this may be clear, but in others identifying the aetiology of PH may require comprehensive and systematic investigation.
In addition to TTE, the typical diagnostic work-up for a patient with PH includes:
- full lung function testing
- imaging for chronic thromboembolic disease (ventilation-perfusion scan or CT pulmonary angiogram)
- imaging for parenchymal lung disease (high-resolution chest CT)
- overnight polysomnography for patients with symptoms of sleep-disordered breathing
- autoimmune serological tests, such as testing for antinuclear antibodies (ANA), antibodies to extractable nuclear antigen (ENA), double-stranded DNA (dsDNA) and anti-cyclic citrullinated peptide (anti-CCP), antineutrophil cytoplasmic antibodies (ANCA) and rheumatoid factor
- HIV serological tests
- abdominal ultrasound examination for portal hypertension.
Right-heart catheterisation
The definitive test for the diagnosis of PH is right-heart catheterisation. This is a safe but invasive test that enables direct measurement of the pulmonary artery pressure, as well as providing the pulmonary artery wedge pressure and an estimate of cardiac output. The pulmonary artery wedge pressure provides an estimate of left atrial pressure and is crucial to determine whether PH is pre-capillary or post-capillary. An elevated pulmonary artery wedge pressure indicates elevated left atrial pressure and suggests that the PH is due to left heart disease. Measurement of cardiac output is essential to derive pulmonary vascular resistance, and catheterisation data that do not include cardiac output are of limited utility. Vasoreactivity testing with a short-acting pulmonary vasodilator (e.g. inhaled nitric oxide) should be performed for patients with idiopathic, heritable and drug-related PAH as this identifies a small (less than 10%) subgroup of patients who may benefit from calcium-channel blocker therapy.
Given the complexity of the diagnostic work-up, it is recommended that patients with significant PH on TTE be referred to a PH specialist centre or, if this is not accessible, to a cardiologist or respiratory physician with experience in this area. A list of approved PAH designated (prescribing) centres is available from the Pulmonary Hypertension Association Australia (www.phaaustralia.com/page/ 20/approved-ph-prescribing-centres).
Management
Over the past 20 years, significant therapeutic progress has been made for patients with PAH (Group 1 PH) and CTEPH (Group 4 PH). However, therapeutic options for patients with PH due to left heart disease (Group 2 PH) and to lung disease (Group 3 PH) remain limited. It is important to emphasise that treatment depends on accurate diagnosis of the underlying aetiology. Management approaches for the different PH groups are summarised in Table 2.
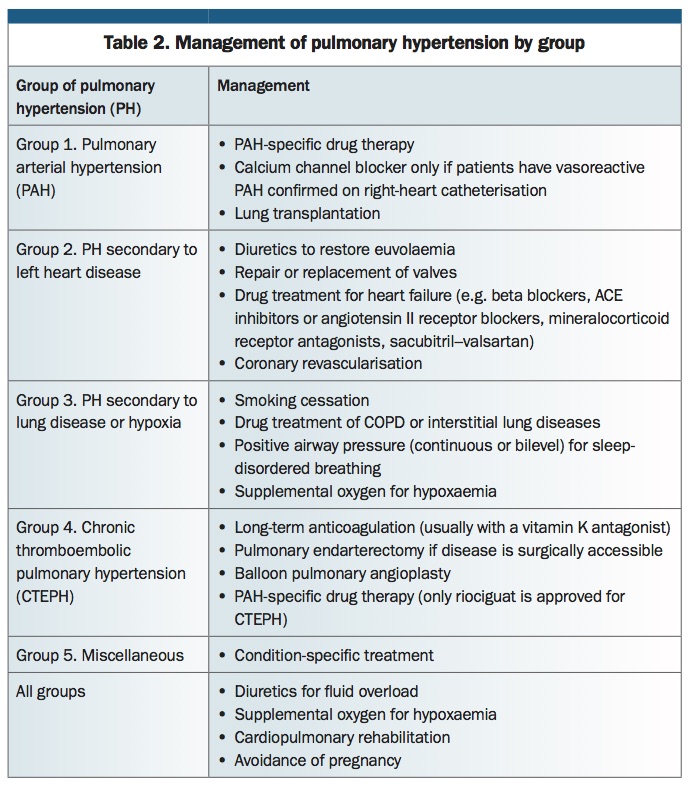{kind=link}
General measures
Close attention to fluid status is important for patients with any type of PH, and diuretics should be used in patients with clinical fluid overload to relieve symptoms. With advanced disease, maintaining the balance between right heart failure, adequate systemic blood pressure and renal perfusion can be challenging; these patients require close clinical and biochemical monitoring.
In addition to medications, nonpharmacological therapies include cardiopulmonary rehabilitation to improve exercise capacity, regular vaccination to prevent infective exacerbations, and supplementary oxygen for hypoxaemia.
Patients with PH have increased perioperative risk, and thus a thorough preoperative assessment by a physician with expertise in managing PH is essential to guide decision-making for patients requiring surgery. For patients with severe PH, the need for elective procedures must be weighed against the risk of major complications. If surgery is indicated then a cardiac anaesthetist and PH specialist should be involved in perioperative management.
Pregnancy is contraindicated for women with severe PH because of the high risk of maternal and fetal mortality. It is important to counsel female patients on these risks and appropriate methods of contraception. The combined oral contraceptive pill should be avoided because of thrombosis risk, but depot contraceptives such as depot etonogestrel and the levonorgestrel-releasing intrauterine system are suitable contraceptive options. Women at risk of a vagal response during insertion of the intrauterine system should undergo this procedure in a hospital gynaecology clinic.
Atrial arrhythmias such as atrial flutter and fibrillation are relatively common in patients with severe PH and may cause cardiac decompensation, particularly with higher heart rates. Haemodynamically unstable arrhythmias require urgent inpatient intervention and attempt at cardioversion.
There is no high-quality evidence to support the use of anticoagulation in all patients with PH. In general, anticoagulation is restricted to those with CTEPH or with concomitant diseases that require anticoagulation (e.g. atrial fibrillation). The benefit of anticoagulation in patients with idiopathic PAH is unclear. Anticoagulation is potentially harmful in patients with PAH associated with systemic sclerosis, as these patients have an increased risk of gastrointestinal tract bleeding.
Group 1. Pulmonary arterial hypertension
Eight different PAH-specific drugs are now approved in Australia and available via the PBS for the treatment of PAH. They are broadly divided into four groups according to their mechanisms of action. These comprise:
- endothelin-1 receptor antagonists (bosentan, macitentan and ambrisentan)
- phosphodiesterase type 5 inhibitors (sildenafil and tadalafil)
- soluble guanylate cyclase stimulators (riociguat)
- prostacyclin derivatives (iloprost and epoprostenol).
These drugs all act as pulmonary vasodilators and exert antiproliferative effects on vascular remodelling (Figure 3). Large, randomised event-driven trials have shown that the PAH drugs improve exercise capacity and long-term outcomes (e.g. they reduce clinical worsening).10,11 More recently, compelling evidence has emerged supporting the use of combination therapy in PAH, in which mechanistically different drugs are used together. Combination therapy is now considered the standard of care in PAH and should be used as initial therapy in most patients, particularly those with severe disease.4
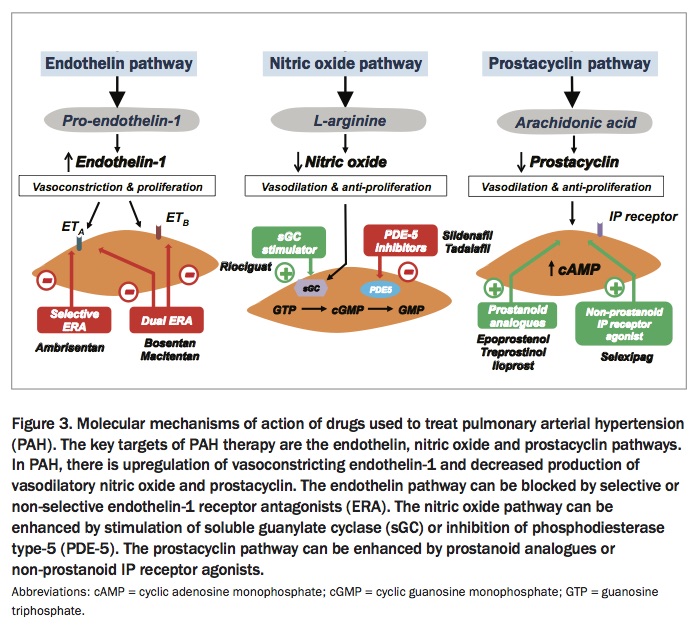{kind=link}
Subsidised access to PAH agents requires diagnostic evaluation and regular follow up at a designated PH prescribing centre. Given the complexity of this process, it is highly recommended that patients are referred to a specialised PH centre as early as possible. At present, the PBS subsidises only one PAH medication at any time. Thus, combination therapy in Australia is accessed via private funding, hospital funding or compassionate access schemes.
Lung transplantation is reserved for patients with severe PAH refractory to maximal medical therapy. Most patients with PAH do not require heart–lung transplantation as the right ventricle recovers its function following bilateral lung transplantation. Data on long-term outcomes of patients who have received a lung transplant for PH estimate survival to be 59% at five years post-transplantation.12
Group 2. PH secondary to left heart disease
Treatment of patients with group 2 PH involves reducing left ventricular filling pressures and afterload by various mechanisms, depending on the underlying aetiology of the left heart dysfunction. Left ventricular filling pressure can be reduced through diuresis to attain euvolaemia, as well as optimisation of heart-failure specific drug therapies for impaired systolic function, including beta blockers and ACE inhibitors.
Stenotic or regurgitant mitral or aortic valve disease may be amenable to surgical or percutaneous valve repair or replacement. Congenital lesions resulting in inflow or outflow tract obstruction, such as occurs in hypertrophic obstructive cardiomyopathy, may similarly be amenable to surgical or percutaneous interventions. Where indicated, coronary revascularisation can improve left ventricular function. Finally, treatment of systemic hypertension to reduce left ventricular afterload reduces left ventricular filling pressures, as well as preventing the subsequent development of left ventricular diastolic dysfunction.
It is important to note that PAH-specific drugs are not approved for use in PH secondary to left heart disease. When combined with elevated left-heart filling pressures, the pulmonary vasodilation and increased pulmonary blood flow that result from their use can potentially precipitate pulmonary oedema and decompensated heart failure. Numerous randomised controlled trials investigating the efficacy and safety of endothelin-receptor antagonists and prostanoids in PH secondary to left heart disease have shown no benefit or even harm, in some cases resulting in premature trial cessation because of increased mortality.13
Group 3. PH due to lung disease or hypoxia
PH secondary to lung disease or chronic hypoxia requires treatment of the underlying condition, including smoking cessation and inhalers for COPD, antifibrotic therapies for pulmonary fibrosis, and positive pressure ventilation for sleep apnoea, as well as correction of hypoxia with supplemental oxygen. Several key trials have shown that oxygen therapy has a benefit in stabilising progression of PH and a mortality benefit in patients with COPD and hypoxaemia.14,15
There are currently no high-quality data to support the use of PAH drugs in PH caused by lung disease. Available data from randomised controlled trials are conflicting, and it is unclear whether there is a relationship between haemodynamic changes, functional parameters and significant clinical improvement.16 Patients with lung disease and severe PH should be referred to an expert centre for evaluation, and enrolment in a clinical trial is encouraged.
Group 4. Chronic thromboembolic pulmonary hypertension
Patients with CTEPH require long-term anticoagulation to prevent further thromboembolism. However, this therapy does not address the fibrotic thrombi that persistently obstruct the pulmonary arterioles and thus may not improve symptoms.
Pulmonary endarterectomy involves removal of the organised thrombi from the pulmonary arteries. It is performed under cardiopulmonary bypass and requires periods of complete circulatory arrest. The procedure is potentially curative, making CTEPH an important diagnosis not to miss. In about 60 to 70% of patients with CTEPH, the disease is amenable to pulmonary endarterectomy. Patient selection remains a complex process based on the anatomical site of disease and surgical expertise.17 All patients with CTEPH should be referred to an expert centre with experience in pulmonary endarterectomy to determine their surgical suitability.
In patients who are deemed not suitable for pulmonary endarterectomy because they have inaccessible distal disease or significant medical comorbidities, other therapies should be considered. Riociguat is listed on the PBS for the treatment of inoperable CTEPH and has been shown to improve exercise capacity.18 Endovascular intervention with pulmonary balloon angioplasty improves haemodynamics and exercise capacity in patients with inoperable CTEPH.19 This relatively new technique is likely to have an expanding role in the management of CTEPH.
Prognostic factors
In general, the prognosis of PH is based on:
- the severity of disease at diagnosis
- rate of disease progression
- response to therapy
- the patient’s overall functional status
- severity of associated comorbidities such as left heart failure or COPD.
The development of right ventricular dysfunction or clinical right heart failure is a poor prognostic sign. Indeed, right ventricular dysfunction is the key determinant of outcome in PH, rather than the absolute level of pulmonary artery pressure.20
Summary
PH encompasses an aetiologically diverse group of conditions that result in elevated pulmonary artery pressure and may ultimately progress to right heart failure. The diagnostic work-up of patients with PH is complex and should be undertaken in specialist PH referral centres. Importantly, accurate classification is crucial for directing management and selection of appropriate therapies. At present, pulmonary vasodilators are indicated only for patients with Group 1 PAH and for those with Group 4 CTEPH with inoperable disease.
High-risk groups, such as patients with connective tissue disorders (e.g. systemic sclerosis), advanced parenchymal lung disease or congenital heart disease, and those with unexplained progressive dyspnoea should be screened for PH with TTE. Early referral is key to the timely investigation and management of PH. CT
References
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.