Hypertrophic cardiomyopathy. Keys to minimising morbidity and mortality
Heart failure
Hypertrophic cardiomyopathy can be an asymptomatic condition or can cause incapacitating symptoms. It is the most common cardiovascular genetic condition and genetic testing of affected individuals and their family members can help prevent the morbidity and mortality associated with hypertrophic cardiomyopathy.
- Hypertrophic cardiomyopathy (HCM) is a primary cardiomyopathy with an abnormally thickened myocardium.
- Patients with HCM display clinical diversity, from no symptoms to heart failure and sudden cardiac death.
- HCM is caused by mutations in genes that encode the sarcomere, the basic contractile unit of the heart.
- Treatment options include lifestyle modififi cations, medical therapies and interventions such as implantable cardioverter defifi brillator therapy and surgical myectomy.
- It is important to clinically screen fifi rst-degree relatives of patients with HCM.
- HCM is best managed in the setting of a specialised multidisciplinary clinic.
Hypertrophic cardiomyopathy (HCM) is a condition of abnormally thickened myocardial muscle in the absence of other conditions that elevate left ventricular afterload, such as hypertension and aortic stenosis. HCM is the most common cardiovascular genetic disorder, with an estimated population prevalence of 0.5% or one in 200 people with pathogenic genetic mutations.1 The classic distribution of hypertrophy in patients with HCM involves the interventricular septum and is known as asymmetric septal hypertrophy; however, other phenotypic variants include concentric, apical and biventricular (also involving the right ventricle) patterns of hypertrophy.
Clinical features
Patients with HCM may be asymptomatic or their symptoms may be incapacitating, with symptom severity related to the severity of left ventricular hypertrophy (LVH), the degree of left ventricular outflow tract obstruction (LVOTO), the amount of mitral regurgitation and the occurrence of atrial or ventricular arrhythmias. Common symptoms include atypical chest pain, dyspnoea and reduced exercise tolerance. Patients may also have symptoms related to pulmonary congestion including orthopnoea and paroxysmal nocturnal dyspnoea, palpitations, presyncope or syncope. Symptoms are often reported to be worse on exertion due to dynamic variations in LVOTO, reduced diastolic filling time due to tachycardia and increased myocardial oxygen demand. Occasionally, the first manifestation of HCM is sudden cardiac death.
Physical examination may provide clues to the presence of HCM and LVOTO. Midsystolic outflow obstruction may result in jerky peripheral and carotid pulses, a double-impulse ‘tapping’ apex beat and a harsh left-sided systolic murmur that is characteristically accentuated on release of the Valsalva manoeuvre. A pansystolic murmur of mitral regurgitation or irregular pulse of atrial fibrillation (AF) may also be detected. If present, clinical features of heart failure are often less marked, although they may include signs of both left heart failure and secondary right heart failure.
Diagnosis
Role of cardiac imaging
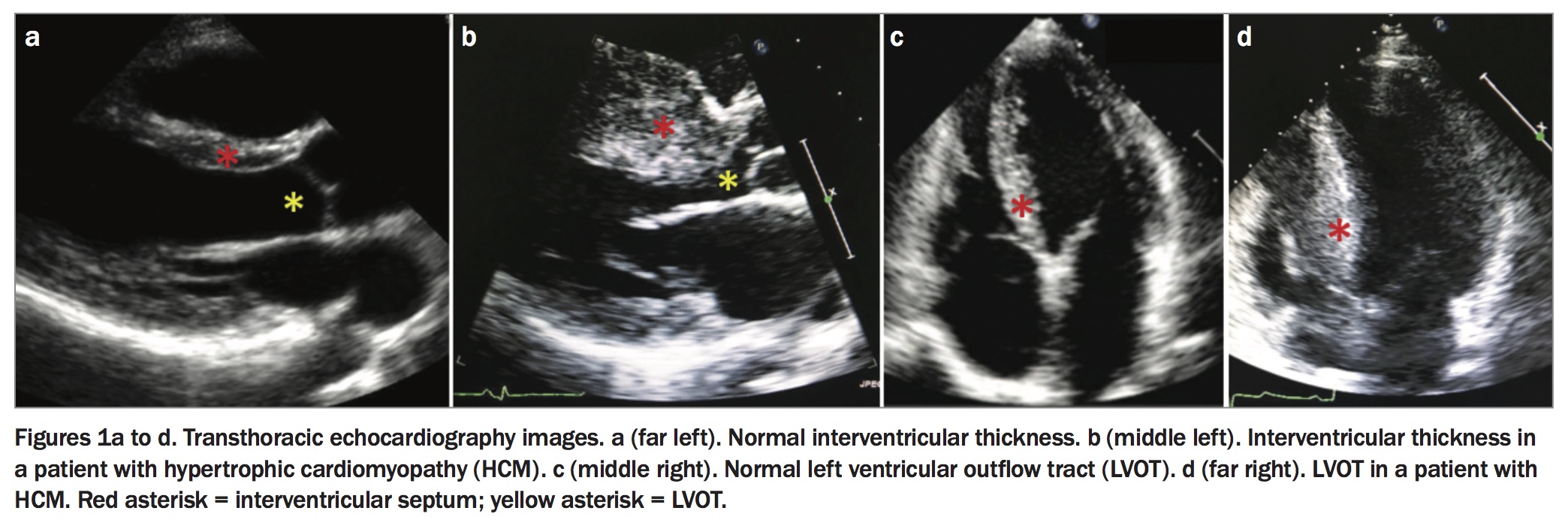
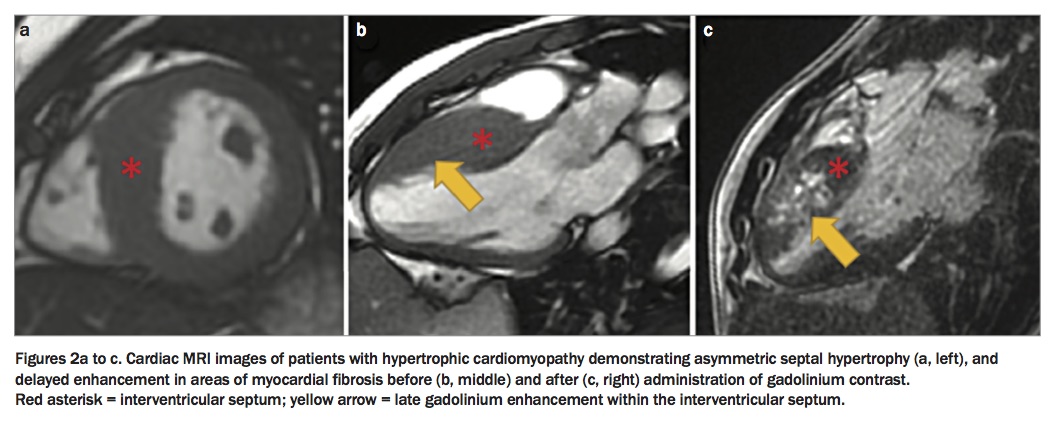
HCM may be diagnosed clinically (by phenotype; Table 1) or genetically (by genotype). A clinical diagnosis requires demonstration of unexplained LVH (≥1 mm) with a nondilated left ventricle on cardiac imaging. Transthoracic echocardiography remains the mainstay imaging technique for diagnosis, quantifying the degree and distribution of LVH, abnormal left ventricular systolic or diastolic function, degree of LVOTO, severity of mitral regurgitation and left atrial size (Figures 1a to d). More recently, cardiac MRI has emerged as an important clinical tool in the diagnosis of HCM, enabling more detailed assessment of hypertrophy and associated morphological anomalies such as aneurysms, and identification of myocardial fibrosis by delayed contrast washout in areas of myocardial scar (late gadolinium enhancement; Figures 2a to c). Fibrosis that is seen in more than 15% of the left ventricle has been associated with a higher risk of sudden cardiac death, although this result needs to be replicated in larger cohorts of patients with HCM.2
{kind=link}
{kind=link}
{kind=link}
Role of genetic testing
Familial HCM is inherited in an autosomal dominant pattern with variable penetration, meaning that an affected person has a 50% chance of passing the gene mutation on to their children, male and female alike. However, the clinical features may vary significantly in severity between family members. The predominant mutations involve genes encoding sarcomere proteins involved in the myocyte contractile apparatus such as myosin-binding protein C (MYBPC3), beta-myosin heavy chain (MYH7), tropomyosin (TPM1) and cardiac troponin T (TNNT2).3 More than 1500 distinct gene mutations have been identified in patients with HCM. Genetic testing of the 10 most common sarcomere genes in patients with HCM results in an overall detection rate of 40 to 50%, which rises to 70 to 80% if there is a positive family history of HCM.4
Clinical screening, including an ECG and echocardiogram, is recommended for all first-degree relatives of patients with HCM.4 Due to the clinical heterogeneity of the condition, a mildly symptomatic proband (the first family member diagnosed with the condition) may have affected relatives with more severe manifestations at younger ages. ‘Cascade testing’ is a sequence of genetic testing in which relatives are tested in a cascading fashion, starting with first-degree relatives and, if indicated due to the presence of severe disease phenotypes, second-degree relatives are then tested. This process enables early diagnosis and appropriate clinical surveillance of
gene-positive individuals or, alternatively, release from lifelong surveillance of
gene-negative family members, and timely institution of potentially life-saving therapies in the setting of high-risk clinical features.
Genetic testing should be undertaken in specialised multidisciplinary centres seeing high volumes of patients (see www.heartregistry.org.au). Such centres must provide a robust genetic counselling service both before and after testing to prepare individuals for the potential results of genetic screening and the implications for their family members.
Other investigations
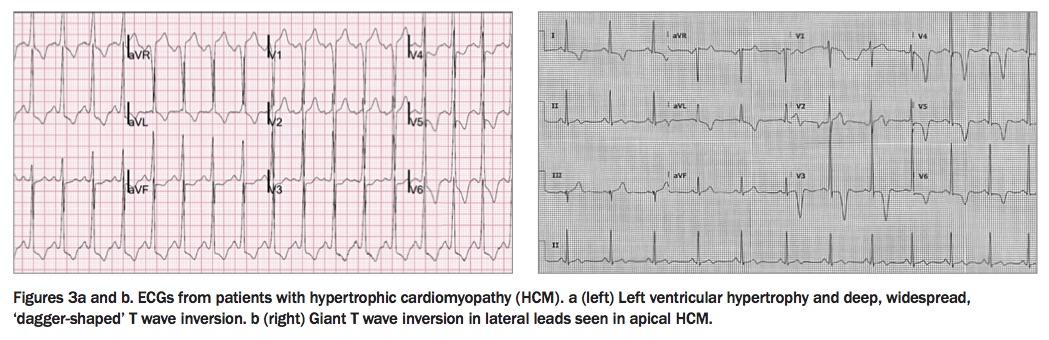
ECG abnormalities that can be seen in patients with HCM include voltage criteria for LVH, ST-segment depression and/or T wave inversion in the left-sided leads, with more marked LVH occasionally demonstrating deep, dagger-shaped T wave inversion (Figure 3a). Giant T wave inversion may indicate an apical form of HCM (Figure 3b).
{kind=link}
Holter monitoring may detect arrhythmias in symptomatic individuals, and may be useful in risk stratification for sudden cardiac death. Stress echocardiographic testing can provide information on dynamic changes in LVOTO and assess for
high-risk features such as exercise-induced blood pressure drop or arrhythmia.
Endomyocardial biopsy may be undertaken in the work-up of HCM to identify infiltrative cardiomyopathies. The pathological hallmark of HCM is disarray of hypertrophied myocytes interspersed with interstitial fibrosis.
Differential diagnoses (HCM phenocopies)
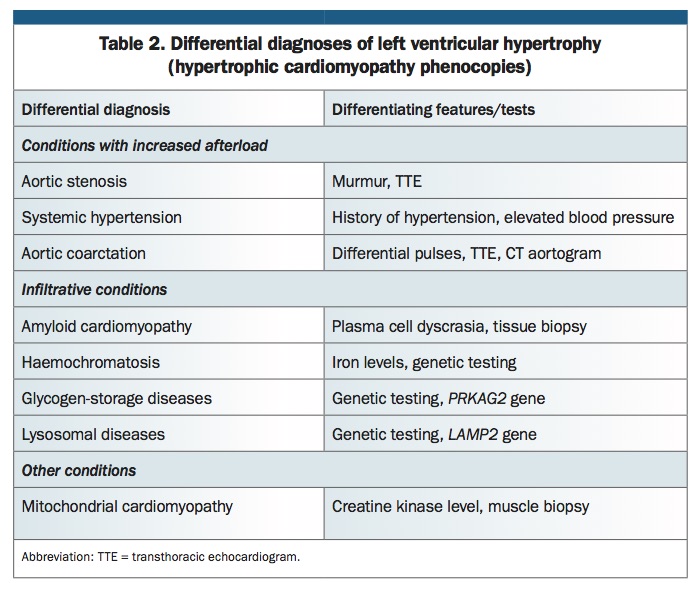
Several conditions can result in hypertrophic myocardial changes that are phenotypically similar to those seen in classic and variant HCM (Table 2). Diagnosis of HCM requires exclusion of other conditions that can lead to LVH such as aortic stenosis, aortic coarctation and hypertension. Less common causes of increased left ventricular mass and wall thickness include the infiltrative cardiomyopathies such as amyloid cardiomyopathy, glycogen-storage and lysosomal diseases (i.e. alpha-galactosidase deficiency in Fabry disease, acid maltase deficiency in Pompe disease, LAMP-2 deficiency in Danon disease, PRKAG2 cardiomyopathy), and mitochondrial cardiomyopathy. Genetic testing can precisely identify some of these HCM phenocopies, which, although rare, represent important diagnoses due to the availability of disease-modifying treatments such as enzyme replacement therapy for Fabry and Pompe diseases.
{kind=link}
Management of HCM
Lifestyle modification
All patients with HCM are advised against taking part in any competitive sports or strenuous exercise (e.g. tennis, sprinting, squash or basketball) because high-level exercise can provoke ventricular arrhythmias and sudden cardiac death. Recent studies suggest that some patients with well-managed HCM and excellent symptomatic control may be able to return to competitive sports under close supervision.5
Pharmacological therapy
Beta blockers, such as metoprolol, are first-line therapy in patients with symptomatic LVOTO to reduce their heart rate and permit more left ventricular filling at rest and with exercise. They can also reduce the severity of LVOTO. Second-line options include disopyramide, which may reduce resting left ventricular outflow tract (LVOT) gradients, and verapamil, which can assist in relieving chest pain in HCM but should be avoided in heart failure with systolic dysfunction.
Pharmacological therapy is not disease modifying in HCM, and there is no evidence that these medications prevent sudden cardiac death in these patients.
Alcohol septal ablation
For patients with persistent symptoms or heart failure despite receiving maximum tolerated pharmacological therapy, alcohol septal ablation or surgical myectomy may be considered. Alcohol septal ablation uses an endovascular approach similar to coronary angiography, in which ethanol is injected into small branch coronary arteries that supply the proximal interventricular septum, causing occlusion and infarction of that coronary territory. As the infarcted area atrophies, LVOTO is relieved. This is a less invasive technique than open myectomy and may be a more favourable option in patients with higher perioperative risk such as older patients with significant comorbidities. However, alcohol septal ablation has a lower success rate than open myectomy at relieving LVOTO and a higher risk of causing high-grade conduction disease requiring pacemaker implantation.
Surgical myectomy
Since first being reported in 1961, surgical myectomy has been the gold standard for treatment of patients with medication-refractory HCM, particularly those with LVOT gradients of more than 50 mmHg who are candidates for open-heart surgery.6 This technique involves direct visualisation of the thickened myocardium and resection of the abnormal tissue, relieving LVOTO, resolving mitral regurgitation and restoring normal physiology. In experienced referral centres, operative mortality is less than 1%. Postoperative complications include bradyarrhythmia requiring permanent pacing, iatrogenic ventricular septal defect, stroke and new AF.7
Atrial fibrillation
Increased left ventricular diastolic pressure and mitral regurgitation increase pressure and volume loading on the left atrium, leading to dilation, fibrosis, abnormal remodelling and ultimately to the development of AF. Patients with HCM often poorly tolerate rapid ventricular rates, as tachycardia shortens diastolic ventricular filling time, increases LVOTO due to reduced diastolic filling and reduces myocardial efficiency. The development of AF in patients with HCM should be treated aggressively with the goal of maintaining sinus rhythm for as long as possible.8 Electrical and pharmacological methods may be used to re-establish and maintain sinus rhythm, and early referral for AF ablation procedures should be considered to restore sinus rhythm more permanently. In the case of established permanent AF, the aim is to achieve effective rate control with conventional agents. Anticoagulation is indicated in all patients with AF and any risk factor for stroke.
Risk stratification for sudden cardiac death and implantable cardioverter defibrillator therapy
Myocardial fibrosis in patients with HCM can act as a substrate for ventricular arrhythmias such as ventricular tachycardia or fibrillation. The most feared complication of HCM is sudden cardiac death, which occurs in about 0.5% of predominantly young to middle-aged patients annually.9 The cause of death is largely due to ventricular arrhythmias, which is why the use of implantable cardioverter defibrillators (ICDs) in HCM has significantly altered the mortality rate of the condition. ICD therapy is the only proven medical strategy to prevent sudden cardiac death in patients with HCM.
Evaluating the risk of sudden cardiac death and deciding which patients should receive primary preventive ICDs is an integral part of the clinical assessment of HCM and is based on the presence of known sudden cardiac death risk factors (Box). Secondary preventive ICDs should be recommended in all patients with HCM and previous aborted sudden cardiac death (i.e. survived cardiac arrest). A risk score calculator that takes several risk variables into account has been developed and recommends consideration of ICD implantation based on the calculated five-year risk of sudden cardiac death (low risk, <4%; intermediate risk, 4 to 6%; high risk, >6%; available at https://qxmd.com/calculate/calculator_303/hcm-risk-scd).10
Patients with one or more high-risk features for sudden cardiac death should have ICD therapy considered. Patients who have other features associated with increased risk of sudden cardiac death should have ICD therapy considered on a case-by-case basis, with the risks and benefits openly discussed with the patient.
Heart failure and transplantation
Heart failure in patients with HCM is due to diastolic dysfunction until late in the disease course when patients may also develop systolic dysfunction. Hypertrophied myocardium is stiffer and has an impaired ability to relax during diastole, meaning that the pressure in the left ventricle is higher than normal throughout the cardiac cycle. This elevated pressure is transmitted to the left atrium causing dilation; in turn it is transmitted through the pulmonary circulation causing pulmonary congestion; subsequently it moves to the right side of the heart leading to right-sided heart failure. Patients with outflow gradients of more than 30 mmHg at rest are at greater risk for HCM-related progressive heart failure or death due to heart failure or stroke.
Heart failure in patients with HCM is managed as per heart failure guidelines based on whether ejection fraction is preserved or reduced. This may additionally involve rate and rhythm control strategies if AF is present. In patients with HCM and treatment-refractory heart failure, heart transplantation is a definitive option for patients younger than 65 years who do not have contraindications such as advanced medical or psychiatric comorbidities or social circumstances preventing treatment adherence. Survival following heart transplant in HCM has been reported at 75% at five years and 60% at 10 years.11
Conclusion
HCM is a clinically heterogeneous genetic disease of wider prevalence than traditionally recognised, and has major implications for not only the affected patient but also their family members. Life expectancy has been significantly improved by:
- advances in diagnosis such as genetic testing and cardiac imaging
- progress in management including ICD therapy
- interventions to relieve LVOTO
- heart transplantation
- development of specialised multidisciplinary clinics with experience in managing patients with this complex
- condition. CT
References
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.