Faint of heart. Syncope and familial long QT syndrome

Congenital heart defects
Hayden is a 13-year-old boy who, accompanied by his concerned mother, presents to your practice after collapsing unconscious while playing soccer. He recovered quickly and felt ne, so his coach had not called an ambulance. Hayden remembers drinking plenty of water before the game. Hayden has no known medical problems and is fully immunised. He denies any episodes of feeling dizzy, but has felt his heart beat forcefully at times. As Hayden is unsure of the duration of these episodes, you ask him to tap on your desk to provide an example. He taps at a rate of well over 100 beats per minute for a period of 15 to 20 seconds. Importantly, Hayden does not consume any caffeine or energy drinks. You ask about any sudden deaths in the family, and his mother reports that her brother died unexpectedly at 34 years of age. She recalls the autopsy was ‘normal’, however you acknowledge that routine genetic testing was not yet available at that time.
What does the clinical examination show?
Hayden looks comfortable at rest. He is 156 cm tall, weighs 52.5 kg and has a body mass index of 21.3 kg/m2, which is within the normal range. His heart rate is 67 beats per minute and regular, and his blood pressure is 110/70 mmHg. He is afebrile with a temperature of 36.8˚C.
Hayden’s peripheries are warm and pulses are equal at radial and femoral arteries. He has no conjunctival pallor. His carotid arteries have no bruits, his heart sounds are dual with subtle splitting of the second heart sound on inspiration, which resolves with expiration. This is a normal finding in a young person. His lung fields are clear to auscultation. His abdomen is soft and nontender, and there is no organomegaly. His blood sugar level is normal.
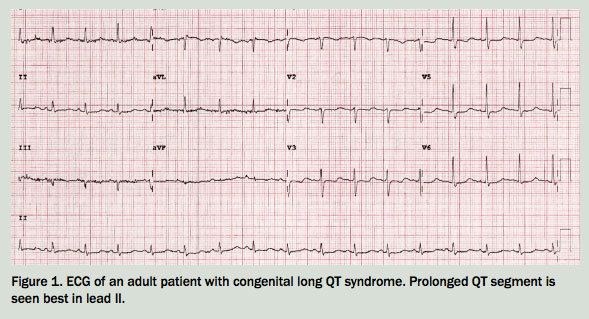{kind=link}
What are the potential causes for this presentation?
The causes of syncope in children are listed in Table 1.1,2 Hayden has exertional syncope in the setting of a family history of probable sudden cardiac death in a second-degree relative. This, along with Hayden’s description of palpitations and the prolonged QT interval on ECG, suggests a cardiogenic cause of syncope. Although further cardiac investigations are indicated, you suspect that he has congenital long QT syndrome (LQTS). Secondary causes for a prolonged QT interval among the general population include electrolyte disturbances, hypothermia, myocardial ischaemia and many medications, including antipsychotics, antiarrhythmics, tricyclic antidepressants and antihistamines. It is unlikely that children will have been prescribed these drugs, but this may be ruled out before investigating further.
{kind=link}
Should Hayden go to hospital?
Features that would require immediate referral to hospital include presyncope or syncope at rest or on minimal exertion, head injury, heart rate or blood pressure outside normal limits and ECG evidence of a ventricular arrhythmia.
How is long QT syndrome diagnosed?
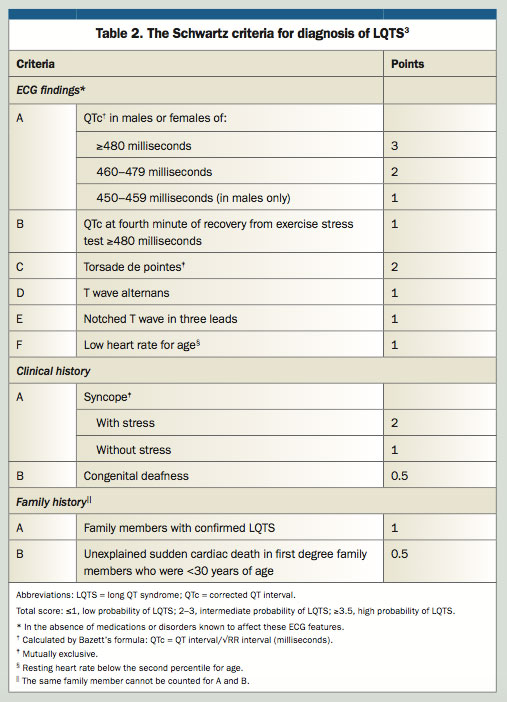
Congenital LQTS is a cardiac channelopathy causing delayed repolarisation of myocardium in a structurally normal heart and carries an increased risk for syncope and sudden cardiac death. The incidence of LQTS is up to one in 2500, and it is most commonly inherited in an autosomal dominant pattern, and very rarely in an autosomal recessive pattern.3 The Schwartz Score (Table 2) provides a scoring system to predict probability of LQTS in patients of all ages, and is based almost completely on the patient’s phenotype.4 The patient’s genotype is also analysed, as LQTS mutations in at least 15 genes have been identified.3 However, the main benefit of genotyping is in the cascade testing of family members once a pathological genotype has been identified. As for any medical procedure, informed consent for genetic testing is given by the individual who is being tested, or their legal guardian. The cost of genetic testing is sometimes covered by clinical genetics and cardiology services, however commercial laboratory testing is not covered by Medicare and may cost in the hundreds to thousands of dollars. Electrophysiological studies are not usually helpful in making the diagnosis.
{kind=link}
Hayden scores five points using the Schwartz scoring system, based on a prolonged QT interval and syncope after physiological stress. This classifies him with a high probability of having LQTS. The second percentile range for heart rate in his age group is 49 to 101 beats per minute at rest.
The Schwartz criteria include congenital deafness due to a rare familial syndrome (Jarvell and Lange-Nielson syndrome) that includes severe sensorineural deafness and LQTS, along with low gastric acid secretion and iron deficiency anaemia.3 The inclusion of family members with confirmed LQTS includes those having received a clinical diagnosis, given not all genes are identified and some diagnoses may predate genetic screening.
How is LQTS managed?
Beta blocker therapy should be initiated in patients with LQTS who have had symptoms, or those with a QTc of >470 milliseconds, particularly in preadolescent boys such as Hayden.3 Metoprolol should not be used. Propranolol is preferred.4,5 In males at high risk due to having the LQT1 genetic subtype, the reduction in fatal arrhythmias by using appropriate beta blockers is 67%.6
In addition, cardiac sympathectomy is a treatment option for patients who have symptoms despite beta-blockade. This involves surgical removal of the first three or four thoracic sympathetic ganglia and results in significant reduction of fibrillation and cardiac events.4
Implantable cardioverter defibrillator (ICD) placement is considered by the cardiologist, based on risk factors and clinical scoring tools. ICDs are recommended in patients who have experienced resuscitated cardiac arrest (strong recommendation), or recurrent arrhythmic syncope while taking beta blockers (moderate recommendation).3 Ultimately, the risk of life-threatening arrhythmia is balanced against the risk of inappropriate shocks, periprocedural or late complications of device and lead placement and the burden of activity limitations that accompany ICD implantation. Subcutaneous ICD systems are preferred in young patients who do not require pacing, as they reduce the complications that arise from transvenous ICD access.3
Triggers for life-threatening arrhythmias can be reduced if patients follow lifestyle guidelines specific to the genetic subtypes of LQTS. In people with the LQT1 subtype, arrhythmias are frequently triggered by exercise or emotional upheaval, and swimming and diving are generally contraindicated. People with the LQT2 subtype are at high risk of arrhythmias if they have derangements of serum potassium levels or experience sudden loud noise, and are advised to avoid loud alarm clocks and telephones. People with the LQT3 subtype can develop arrhythmias at rest or in their sleep, and are advised not to sleep alone or to have an intercom system to detect changes in breathing.5 LQT1, LQT2 and LQT3 together account for up to 85% of cases of familial LQTS.
An adult patient with a similar presentation to Hayden would be investigated and managed in the same manner, and warrants referral to cardiology and genetic services. Overall, LQTS is the most common of the cardiac channelopathies. Others include Brugada syndrome, short QT syndrome and catecholaminergic polymorphic ventricular tachycardia.7 All can result in sudden cardiac death, and if suspected the patient should be referred urgently for further investigation.
References
- Driscoll DJ, Jacobsen SJ, Porter CJ, Wollan PC. Syncope in children and adolescents. J Am Coll Cardiol 1997; 29: 1039-1045.
- Massin MM, Bourguignont A, Coremans C, Comté L, Lepage P, Gérard P. Syncope in pediatric patients presenting to an emergency department. J Pediatr 2004; 145: 223-228.
- Waddell-Smith KE, Skinner JR; members of the CSANZ Genetics Council Writing Group. Update on the diagnosis and management of familial long QT syndrome. Heart Lung Circ 2016; 25: 769-776.
- Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol 2012; 5: 868-877.
- Schwartz PJ. The congenital long QT syndromes from genotype to phenotype: clinical implications. J Int Med 2006; 259: 39-47.
- Goldenberg I, Bradley J, Moss A, et al; International LQTS Registry Investigators. Beta blocker efficacy in high-risk patients with the congenital long-QT syndrome types 1 and 2: implications for patient management. J Cardiovasc Electrophysiol 2010; 21: 893-901.
- Behere SP, Weindling SN. Inherited arrhythmias: the cardiac channelopathies. Ann Pediatr Cardiol 2015; 8: 210-220.
- Australian Genetic Heart Disease Registry. Cardiac genetic services in Australia and New Zealand. Available online at: www.heartregistry.org.au/patients-families/cardiac-genetic-services (accessed June 2017).
Single article purchases are temporarily unavailable due to site maintenance.
If you would like to purchase an article during this time, please email us at [email protected] with the article details and we'll assist you directly. We'll also let you know when online purchasing is available again.
Thank you for your patience and understanding.